Biological Electron Transfer
During my PhD studies and following years at the University of Arizona, I carried out X-ray structural studies of proteins involved in biological electron transfer. This in turn lead to docking studies and other simulations aimed at understanding how electron transfer processes are controlled and facilitated.
CYTOCHROME STRUCTURE
Cytochrome Structural Conservation
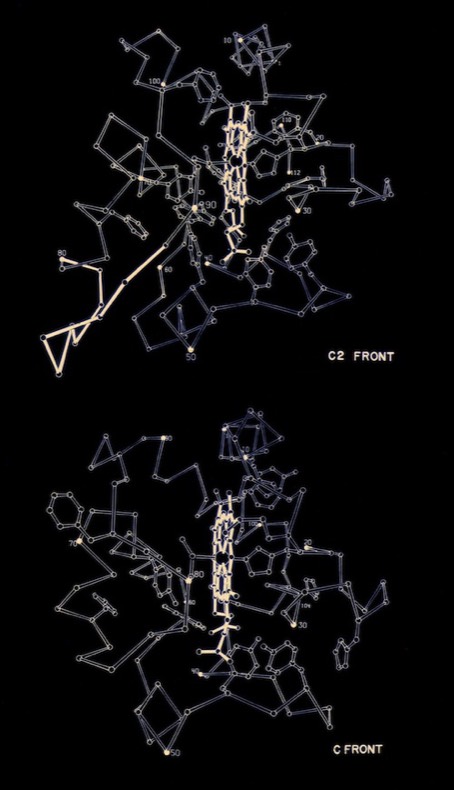
I began PhD studies in Chemistry at UCSD in 1968 where I joined Joe Kraut’s lab. At the time he was collaborating with Martin Kamen who was isolating and characterizing numerous redox proteins from photosynthetic bacteria. I began working to crystallize these proteins, leading to the determination of the structure of cytochrome c2 from the photosynthetic bacterium R. rubrum for my PhD thesis. According to the PDB Newsletter of March 1978, cytochrome c2 coordinates were the third or fourth set deposited at the PDB (there were ~ 20 3D protein structures known at that time), and among the first at the then high resolution of 2 Angstroms.
The ubiquity of cytochromes c among many biological phyla had already made them the subject of numerous studies of protein evolution based on comparative sequence analysis. With the availability of the horse heart cytochrome c structure from the Dickerson lab at CalTech, it became possible, through analysis of conserved features, to address many structure-function questions concerning cytochrome function and mechanism.
The ubiquity of cytochromes c among many biological phyla had already made them the subject of numerous studies of protein evolution based on comparative sequence analysis. With the availability of the horse heart cytochrome c structure from the Dickerson lab at CalTech, it became possible, through analysis of conserved features, to address many structure-function questions concerning cytochrome function and mechanism.
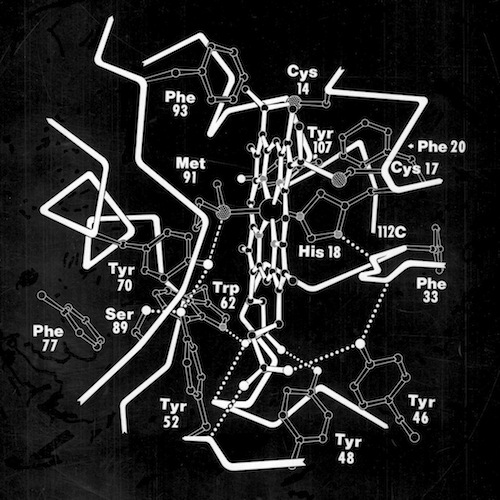
Many questions regarding electron transfer mechanism and redox potential regulation centered on amino acid residues that were conserved among the mitochondrial cytochromes c and the photosynthetic versions like cytochrome c2 and cytochrome c550. These included a PHE residue adjacent the heme (PHE 93 in cytochrome c2) and several TYR and TRP residues that formed H-Bond interactions with heme propionic acid side chains. In the initial, pre-refined structure of cytochrome c2, we interpreted the MET 91 axial ligand density as bonding the heme iron slightly “off axis”. This feature, conjectured to effect redox potential, ultimately refined to a straight axial ligand orientation when subsequently refined with higher resolution data by Barry Finzel. Barry subsequently worked with me both at Genex and DuPont CRD.
The ring of charged residues on the “front” surface of cytochromes c facilitates electrostatic interaction with redox partners and restricts diffusion on negatively charged membrane surfaces with imbedded redox partners to enhance overall electron transfer rates. This mode of interaction implicitly assumes that both heme oxidation and reduction occur through “front-side interactions” although other mechanisms were also proposed that involved alternative pathways.
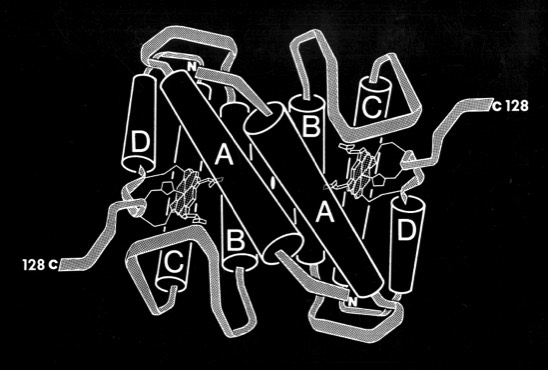
In 1973, I joined the Chemistry faculty at the University of Arizona in Tucson and began a collaboration with Mike Cusanovich. Mike was a former student of Martin Kamen who had expanded on Martin’s work investigating redox proteins in photosynthetic bacteria. Mike was working on a new family of cytochromes that had covalently bound heme groups like conventional c-type cytochromes, but were otherwise sequentially and biophysically quite different. These proteins are widely distributed in photosynthetic bacteria and were termed cytochrome c’ (c-prime).
Pat Weber, who was working with me at the time, carried out the structure determination and found that cytochrome c-prime formed a dimer where each subunit was organized as a 4-alpha helical bundle. The structure determination initiated comparative studies of proteins with the “4-alpha helical bundle” structural motif.
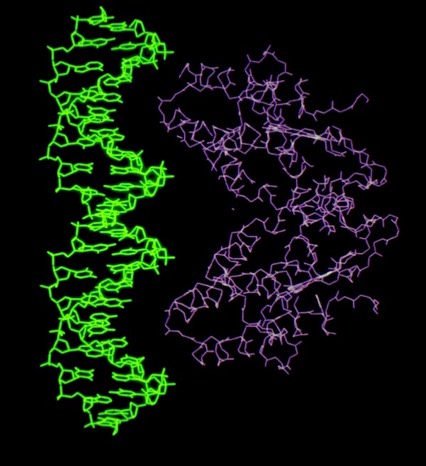
Despite the subsequent X-ray structure determination of several cytochromes c-prime from photosynthetic bacteria, its exact biochemical function (although assumed to involve electron transfer) remains unknown. One interesting possibility we noted is that it makes a highly complementary fit to DNA, which could imply some some redox dependent regulatory function.
PROTEIN DYNAMICS & REDOX POTENTIAL REGULATION
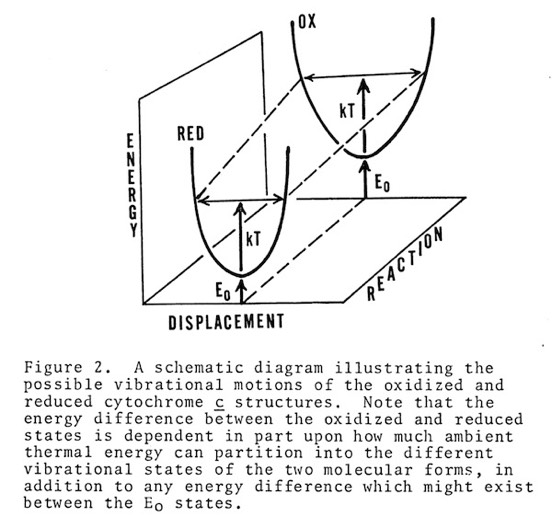
Cytochrome c manifests many biophysical characteristics suggesting that it undergoes a redox-coupled conformational change. Although NMR experiments have subsequently elucidated this transition in detail, early structural studies, that showed the oxidized and reduced structures of cytochromes c and c2 to be essentially identical, ran contrary to some data and expectation that had suggested a change in heme axial iron ligation on reduction. I consequently proposed that the difference, manifest in such phenomena as thermal stability and H-ion exchange, reflected a difference in the dynamic state of the oxidized and reduced structures, and that this could also serve as a way of regulating protein redox potential.
ELECTRON TRANSFER MECHANISMS
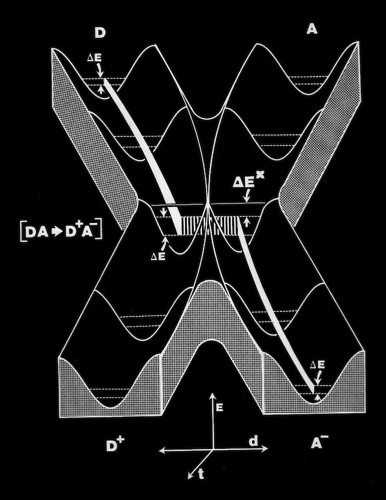
As the first X-ray structures of electron transfer proteins emerged, questions arose regarding the possibility of electron transfer between protein prosthetic groups via classical outer-sphere electron transfer mechanisms. Outer-sphere electron transfer mechanisms were generally envisioned to occur through direct overlap between delocalized orbitals of the donor and acceptor prosthetic groups.
Alternately, John Hopfield proposed that electron transfer could take place via a tunneling mechanism. This required proper relative orientation between the orbital of the donor and acceptor prosthetic groups, but could allow electron transfer to occur at reasonable rates at inter-prosthetic group distances of 8 Angstroms or less. Subsequent modeling studies of the cytochrome c-cytochrome b5 interaction showed that it was not possible to bring the cytochrome heme groups into physical contact, supporting the tunneling mechanism hypothesis for inter-heme electron transfer.
Alternately, John Hopfield proposed that electron transfer could take place via a tunneling mechanism. This required proper relative orientation between the orbital of the donor and acceptor prosthetic groups, but could allow electron transfer to occur at reasonable rates at inter-prosthetic group distances of 8 Angstroms or less. Subsequent modeling studies of the cytochrome c-cytochrome b5 interaction showed that it was not possible to bring the cytochrome heme groups into physical contact, supporting the tunneling mechanism hypothesis for inter-heme electron transfer.
CYTOCHROME C- CYTOCHROME B5 INTERACTIONS
Modeling the Cytochrome c - Cytochrome b5 Reaction Complex
When the structure of Cytochrome b5 became available from Scott Matthews’ lab, it was noted that it also incorporated a ring of charged residues around the accessible surface of its non-covalently bound heme group. However, in contrast to c-type cytochromes, the charge necklace was composed of acidic residues. Although not known to be natural physiological partners, cytochrome b5 and cytochrome c were known to undergo intermolecular electron transfer rates as fast as many known physiological redox partners. In addition, reaction rates were observed to be highly ionic strength dependent, consistent with electrostatic facilitation of the ET interaction.
Coincident with the structure determination of several redox proteins, there was substantial activity in the theoretical community regarding the actual mechanism of electron transfer between proteins. A specific question concerned whether ET took place through outer sphere reactions that required direct orbital overlap between donor and acceptor heme prosthetic groups; or alternatively, that ET could occur through a short-range tunneling mechanism, estimated to be effective over barriers less than 8 Angstroms or so.
Coincident with the structure determination of several redox proteins, there was substantial activity in the theoretical community regarding the actual mechanism of electron transfer between proteins. A specific question concerned whether ET took place through outer sphere reactions that required direct orbital overlap between donor and acceptor heme prosthetic groups; or alternatively, that ET could occur through a short-range tunneling mechanism, estimated to be effective over barriers less than 8 Angstroms or so.
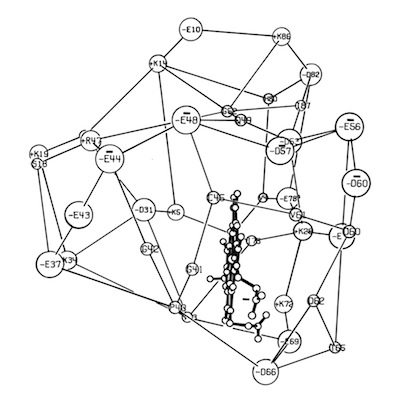
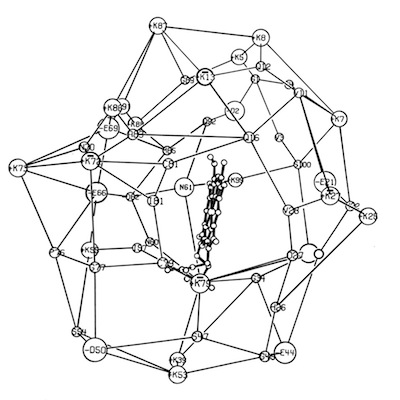
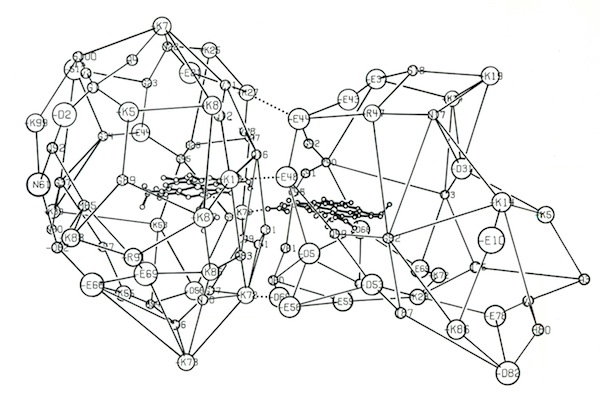
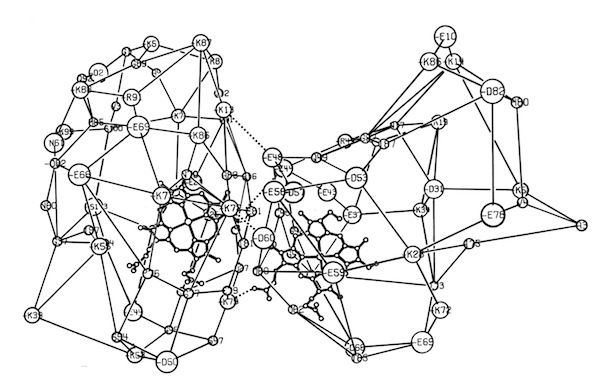
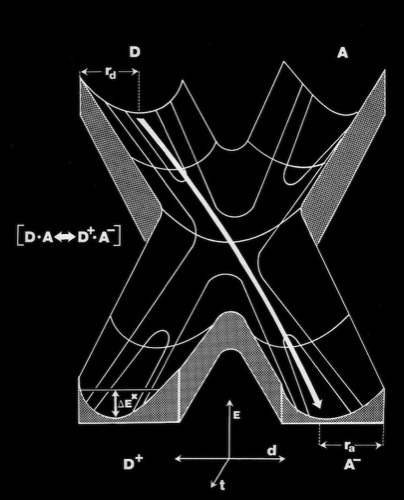
I decided to make a model of the complex formed between Cytochromes c and b5 that would be optimal for ET. The characteristics of this complex would include optimized charge complementarity between positively and negatively charged groups on the cytochrome surfaces together with closest approach and coplanar alignment of the two heme groups. At the time (1975) molecular computer graphics were not generally available. Consequently, to produce the complex geometry, I defined the surfaces of the two molecules as polyhedra (illustrated in 3D using ORTEP at left) with apices defined by “extended” surface charge residues. Following visual alignment of corresponding complimentary charge residues, I was then able to apply a least squares fitting procedure to produce a complex coordinate set which could then be examined for detail fit, charge complementarity, and potential steric interference. After a few iterations involving adjustments consistent with the flexibility of lysine side chains, I achieved a final best model. It turned out that the heme groups could not directly interact but were indeed separated by about 8 Angstroms, consistent with a short range tunneling ET mechanism. This cytochrome c – cytochrome b5 complex model was among the first intermolecular protein docking studies. Many subsequent site-specific modification and chemical modification studies confirmed proposed features of the complex.
Modeling the Molecular Dynamics of the Cytochrome c - Cytochrome b5 Reaction Complex
In 1985 I joined DuPont Central Research, where I was able to assemble a team of outstanding colleagues giving the group broad capabilities in x-ray crystallography and computational chemistry. The DuPont environment was unusual in that we had extraordinary access to state-of-the-art computing resources including CRAY supercomputers, a substantial DEC VAX system, and a STAR ST100 Array Processor. This enabled us to try many sorts of computational and simulation experiments as outlined in Modeling & Simulation.
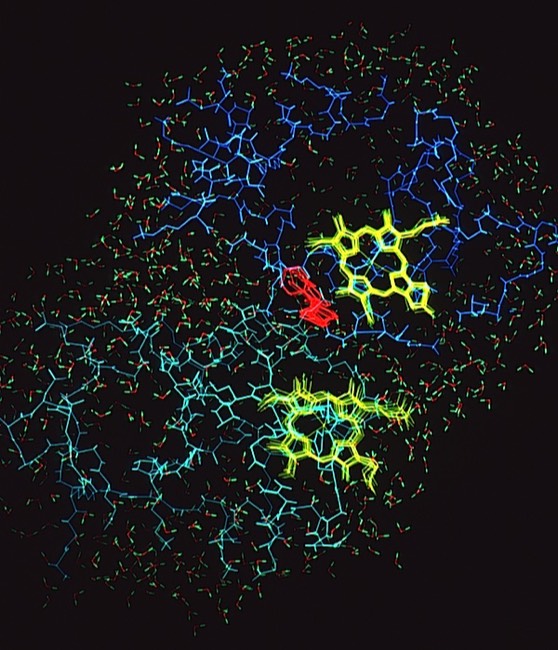
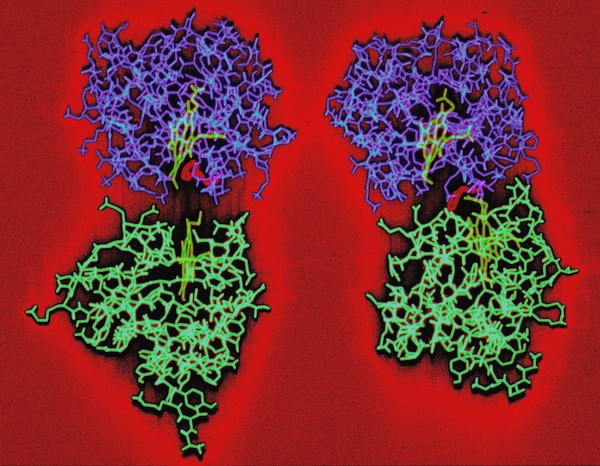
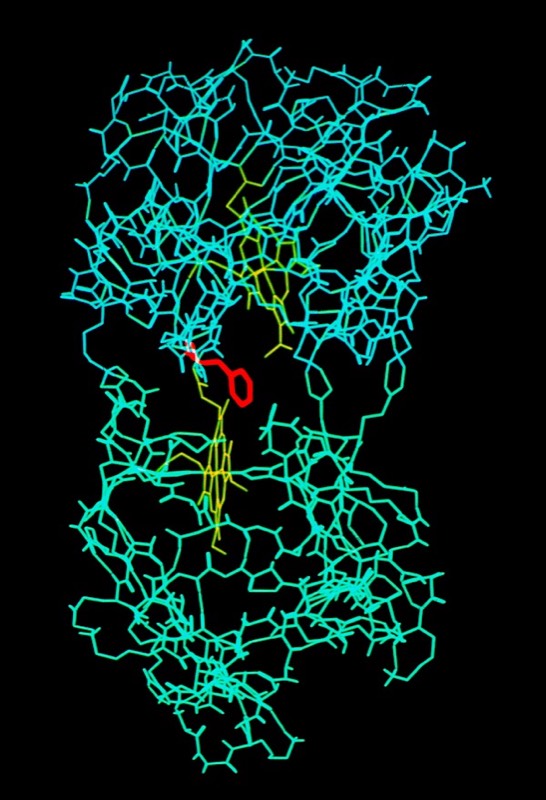
One project that was spearheaded by John Wendoloski revisited the cytochrome c-cytochrome b5 electron transfer complex using molecular dynamics. Simulations were run on DuPont’s CRAY 1A and our groups’ STAR ST100 array processor.
The final run included a fully hydrated complex including both proteins and a 6 Angstrom hydration shell of 1222 TIP3P water molecules. The dynamics were followed for 140 picoseconds and illustrated the considerable flexibility in the relative motions of the two cytochromes that were accessible in an electrostatically docked complex.
In addition, Phe 82, a sequentially invariant aromatic residue packed adjacent the heme in cytochrome c, was observed to sample conformations where it flipped out, potentially forming an electron transfer bridge between the cytochrome heme groups.
CYTOCHROME C - FLAVODOXIN INTERACTIONS
In the early 1980’s, we began to investigate the generality of complementary charge interactions and the role of electroststics in facilitating reversible electron transfer reactions between proteins. The structure of flavodoxin had been determined in Martha Ludwig’s laboratory in 1974. An interesting feature of the structure was the presence of a negative charge necklace around the perimeter the flavin group, one edge of which was exposed on the flavodoxin molecular surface. Gordon Tollin, a colleague at the University of Arizona, was an expert in flavin protein reaction kinetics, and so we formed a collaboration to experimentally investigate the potential role of electrostatics in facilitating interactions between flavodoxin and cytochrome c as redox partners. This work produced two publications.
Cytochrome c- Flavodoxin Biochemical Experiments
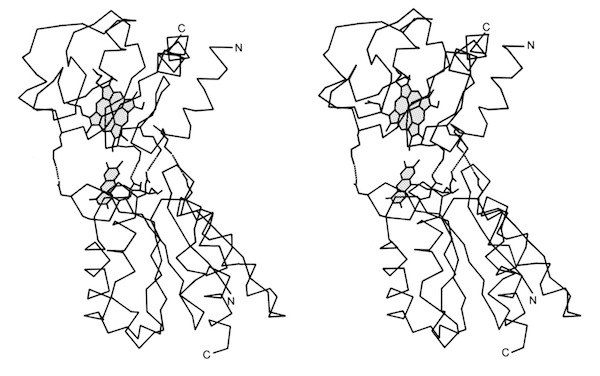
Cytochrome c- Flavodoxin Electrostatic Field Computations
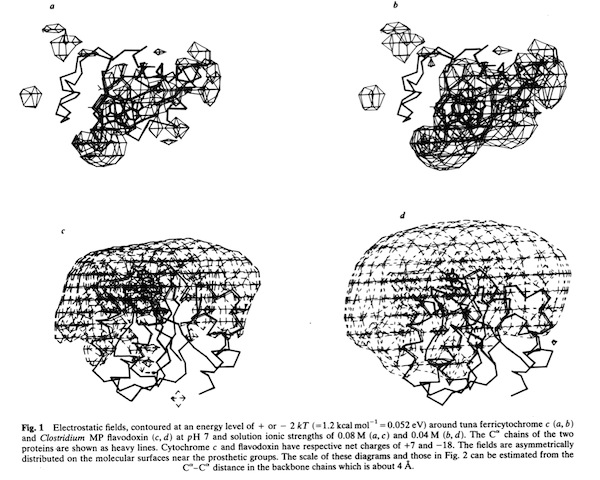
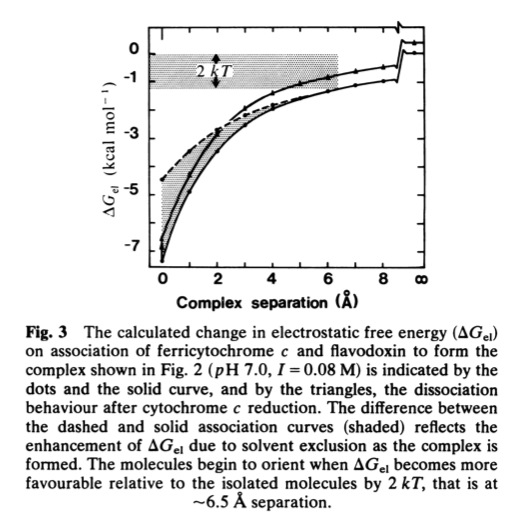

The second, complementary paper, was a purely computational study of the flavodoxin-cytochrome electrostatic field interaction. This work arose from a collaboration with Jim Matthew who was a postdoc with Fred Richards while I was on sabbatical at Yale in 1981. Jim had developed a computational method to represent the electrostatic fields generated by the surface charge distributions on proteins. Although the method involved some geometrical approximations that were ultimately superseded by more extensive calculations, these had little practical effect on estimating field intensities a few Angstroms from the protein surface. In addition, the method readily accommodated the calculation of the electrostatic field dependence on solution ionic strength and pH, thus making it possible to directly couple computational results with experimental measurements of the dependence of ET rates on solution ionic strength.